
Case Published: February 2020
Diagnosis: C3 Glomerulonephritis (C3GN)
Case Summary: Great work! Let’s review this case.
In this case, we have a patient with asymptomatic hematuria. The first step in the evaluation of hematuria is confirmation of hematuria with a repeat urinalysis, followed by investigation of whether the hematuria originates from the glomerulus (suggesting a glomerulonephritis) or elsewhere in the genitourinary tract (i.e. renal cell carcinoma, bladder cancer, vascular disorders, hemorrhagic cystitis, bleeding disorders). The presence of dysmorphic RBCs in more than 25% of the high power (hpf) microscope field is specific, though not sensitive, for glomerulonephritis. In other words, the absence of dysmorphic RBCs does not “rule out” glomerulonephritis, but their presence makes us fairly confident this is a glomerular issue. Glomerular diseases also often present with both hematuria and proteinuria (as in our patient), rather than hematuria alone.
In this case, we did see acanthocytes (a type of dysmorphic red blood cell) under the microscope. Check out this Renal Fellow Network post on acanthocytes. Acanthocytes are pathognomonic for glomerular disease. Take a look at 2 acanthocytes below and notice the “blebs.”

Now that we are looking for a glomerulonephritis…let’s think about our differential. We’re probably looking at a nephritic syndrome, and etiologies of nephrotic syndrome move further down on our differential diagnosis (i.e. membranous nephropathy, HIV nephropathy, diabetic nephropathy, focal segmental glomerulosclerosis).
*Systemic lupus erythematosus (SLE) nephritis
*Post-infectious glomerulonephritis (PIGN)
*Anti-neutrophil cytoplasmic antibody (ANCA) -associated glomerulonephritis
*Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome
*Thrombotic micoangiopathy (TMA)
*Membranoproliferative glomerulonephritis (MPGN)
Our additional diagnostic testing reveals a low C3 and normal C4 – without detection of ANCAs or anti-nuclear antibody (ANA) titers. A low C3 with a normal C4 suggest activation of the alternative complement pathway. When hypocomplementemia is present, try our mnemonic CHAMPS. Without our data thus far, PIGN (though no clinical history of infection) and MPGN rise to the top of the differential. Remember that MPGN is a glomerular injury pattern that can be due to various etiologies including C3 glomerulopathy (C3 glomerulonephritis, dense deposit disease, or CHFR5 nephropathy), infections including hepatitis C virus and hepatitis B virus, autoimmune diseases, C4 glomerulopathy, and monoclonal gammopathies.

Ultimately a kidney biopsy is needed to make the diagnosis. The light microscopy slides are full of findings: thickened glomerular basement membrane, mesangial expansion, double contours, and glomerular lobulation (caused by endocapillary proliferation). These features are consistent with a pattern of membranoproliferative glomerulonephritis (MPGN).

So what are double contours or “tram-tracking”? On the Periodic-Acid Schiff (PAS) stain above, the areas with double contours appear to be white – or areas within the basement membrane that do not stain. This splitting of the basement occurs as a result of mesangial cell interposition and the formation of new basement membrane which is a response to subendothelial immune complex deposits.
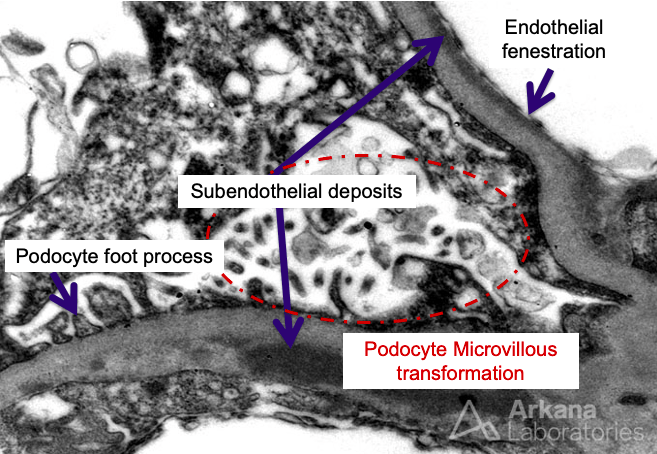
In addition to subendothelial deposits on the electron microscopy (EM), we also see microvillous transformation of the podocytes – which is a fancy way to describe cytoplasmic podocyte extrusions that may indicate podocyte stress. Whenever we find deposits under EM, our next question should be…what are they? The immunofluorescence (IF) often has the answers. Our IF here stains dominantly for C3, but not really much staining of the immunoglobulins or C1q. Thus our diagnosis here is most likely C3 glomerulonephritis (C3GN). Deposits in PIGN are classical more “hump-like.” Deposits in dense deposit disease are…well…dense, ribbon-like deposits along the GBM, tubules, and Bowman’s capsule. Mesangial deposits are also often seen with C3GN.
Patients with C3GN have overactivation of the alternative complement pathway. Below is a figure showing all three complement pathways from Nature Immunology.

Abnormalties in the alternative pathway components above can lead to excessive activation as is observed in patients with C3GN.
*Low or absent Factor H
*Complement factor H-related (CFHR) protein gene mutations
*Deficiency of serum factors B, factor I or membrane cofactor protein (MCP, CD46)
*Gene variants variants of C3 convertase or other complement regulator genes (e.g decay accelerating factor)
Finally, a brief word on treatment– treatment entails various modes of immunosuppresion including complement blockade, corticosteroids, rituximab, mycophenolate mofteil, and calcineurin inhibitors. C3 glomerulopathies are a complex group of diseases – for more, check out the references below:
- Smith RH et al. C3 glomerulopathy – understanding a rare complement-driven renal disease. Nat Rev. Nephrol 2019.
- Bomback AS, Santoriello D, Avasare RS, et al. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int 2018; 93:977.
- Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis–a new look at an old entity. N Engl J Med 2012; 366:1119.
Case Published: May 2020
Case 43 Index
Case 43 Introduction
Case 43 Physical Exam
Case 43 Diagnostic Testing
Case 43 Pathology
Case 43 Additional Pathology
NephSim