
Case 45 Index
Case Published: July 2020
Case Submitted By: Jamie Willows
Diagnosis: Gitelman Syndrome
Case Summary: Strong work! In this case, we’re presented with a patient with hypokalemia and a metabolic alkalosis (pH 7.5 = alkalemia; HCO3 36 meq/L; PCO2 48 mm Hg; for more, check out our Acid-Base tutorial). Multiple mechanisms contribute to the generation and maintenance metabolic alkalosis – check out this review of the pathophysiology of metabolic alkalosis here. The differential diagnosis for metabolic alkalosis can be broken down by volume status: volume depletion vs volume expansion. So in this case, which is it?
Here, we find a hypovolemic patient as evidenced by a relatively low blood pressure, tachycardia, and dry mucous membranes on examination. The next step to identify the etiology is to check the urine chloride (i.e. is there salt wasting?). Wait – why not the urine sodium, as we’ve done in the past to evaluate a patient’s effective circulating volume (ECV)? With increased bicarbonaturia (as is the case with a metabolic alkalosis and increased serum bicarbonate), sodium will be excreted along with bicarbonate in the urine. Thus, it becomes a less reliable marker of ECV – and we turn to our friend chloride.
 In our case with a hypovolemic patient with metabolic alkalosis, urine chloride is high ( > 20 meq/L) – suggesting salt-wasting. So our differential has now been narrowed down to either Bartter syndrome, Gitelman syndrome, or diuretic use. The high urine potassium ( > 15 meq/L) in the setting of hypokalemia suggests potassium-wasting – consistent with these etiologies.
In our case with a hypovolemic patient with metabolic alkalosis, urine chloride is high ( > 20 meq/L) – suggesting salt-wasting. So our differential has now been narrowed down to either Bartter syndrome, Gitelman syndrome, or diuretic use. The high urine potassium ( > 15 meq/L) in the setting of hypokalemia suggests potassium-wasting – consistent with these etiologies.
Our diuretic screen is negative, so we’re left with Bartter vs Gitelman. Bartter syndrome (there are 5 types!) mimics use of loop diuretic while Gitleman mimics use of a thiazide diuretic. How do we tell the difference? Urine calcium! Remember that thiazide diuretics increase calcium reabsorption while loop diuretics promote urinary calcium excertion. So the money test here is quantification of urinary calcium, either using a spot calcium:Cr ratio or a 24 hour urinary calcium (< 300 mg is considered “low”). The low spot Ca:Cr ratio suggests a diagnosis of Gitelman syndrome here.
Gitelman syndrome (GS, first described in 1966 by Gitelman HJ et al.) is an autosomal recessive rare salt-wasting nephropathy that is due to a sodium-chloride symporter (NCC) mutation in the distal convoluted tubule (usually a mutation in the SCL12A3 gene, less commonly in the CLCNKB gene). Non-specific symptoms as in this case are typical, such as fatigue, muscle weakness, nocturia, cramps or palpitations, and patients often report thirst and salt-hunger. However, the phenotype is variable even within families. Many patients are not diagnosed until adulthood and there are likely many undiagnosed cases within the population.
In addition to hypokalemia, hypomagnesaemia due to magnesium wasting in the urine is common but not always present. Proton pump inhibitors may cause hypomagnesemia due to effects on TRMP6/7 and magnseium reabsorption, though would be unlikely to cause the other findings in this case including potassium and salt-wasting. Liddle syndrome and primary hyperaldosternism are unlikely here, given hypotension (rather than hypertension).
There is no cure for GS, and the mainstay of treatment is sodium, potassium and magnesium replacement. There is consensus agreement to target potassium > 3 meq/L and magnesium > 0.6 meq/L, though relief of symptoms should be the goal rather than fixation on strict numerical values.
One randomized control trial has studied treatments in GS, in which use of non-steroidal anti-inflammatory drugs, amiloride or eplerenone to improve serum potassium was hampered by the side-effect profiles of these medications. Information about the long-term sequelae of GS is lacking. GS has been associated with increased rates of sudden cardiac death as well as overall decreased quality of life.
Finally, here’s a table from Renal Fellow Network that summarizes heritable blood pressure disorders:
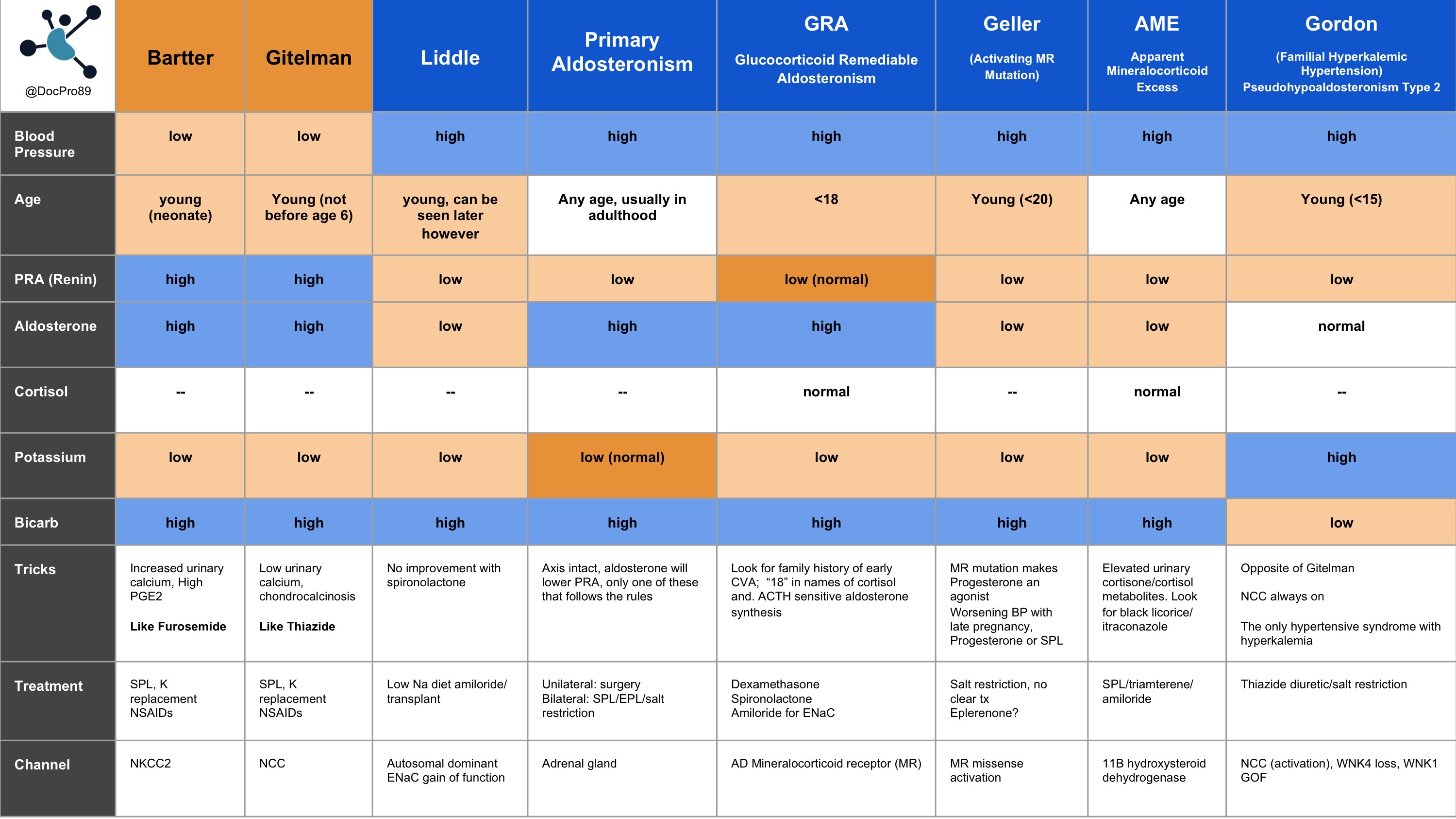
Want to take a deeper dive into GItelman syndrome? Check these out:
3.Pachulski RT, Lopez F, Sharaf R. Gitelman’s not‐so‐benign syndrome. N Engl J Med. 2005;353:850‐851.
Case 45 Index
Case 45 Introduction
Case 45 Physical Exam
Case 45 Diagnostic Testing
NephSIM